MIT : l'IA qui conçoit des médicaments à partir de protéines
Des chercheurs du MIT ont développé un modèle génératif capable de prédire comment des protéines synthétiques interagissent avec des cibles biologiques. Une avancée qui pourrait réduire le temps de développement médicamenteux de dix à deux ans.
Marvin Laurac
Veille technologique
Le développement d'un médicament prend en moyenne douze ans et coûte entre un et trois milliards de dollars. L'étape la plus longue est la phase de découverte : identifier parmi des milliards de molécules candidates celle dont la structure interagira précisément avec la cible thérapeutique souhaitée. C'est un problème combinatoire d'une complexité vertigineuse. Le modèle du MIT s'y attaque directement.
Le problème du repliement des protéines
Pour comprendre l'enjeu, il faut saisir ce qu'est une protéine dans ce contexte. Une protéine est une chaîne d'acides aminés qui se replie dans l'espace tridimensionnel pour former une forme spécifique. C'est cette forme 3D qui détermine ses propriétés biologiques : avec quelles autres molécules elle interagit, à quelle vitesse, et avec quelle affinité.
AlphaFold de DeepMind avait déjà résolu en grande partie le problème de la prédiction du repliement : donner une séquence, prédire la structure 3D. Le MIT va plus loin : donner une cible thérapeutique et une interaction souhaitée, générer une protéine synthétique dont la structure produira cette interaction. C'est le problème inverse, et il est significativement plus difficile.
“Nous ne cherchons plus une aiguille dans une botte de foin. Nous redessinons l'aiguille pour qu'elle s'adapte parfaitement au trou qu'on veut traverser.”
Comment le modèle fonctionne
Le modèle est un diffusion model adapté aux données structurales de protéines. Il est entraîné sur 340 millions de structures protéiques documentées, avec leurs séquences d'acides aminés, leurs formes tridimensionnelles et leurs interactions mesurées expérimentalement. Le modèle apprend la distribution statistique des structures qui produisent des interactions spécifiques.
En inférence, on lui soumet une cible thérapeutique et une description de l'interaction souhaitée. Le modèle génère 100 à 1000 séquences candidates, classées par probabilité estimée d'efficacité. Les chercheurs ne synthétisent ensuite que les cinq à dix meilleures candidates pour les tests expérimentaux.
Résultats préliminaires sur le cancer
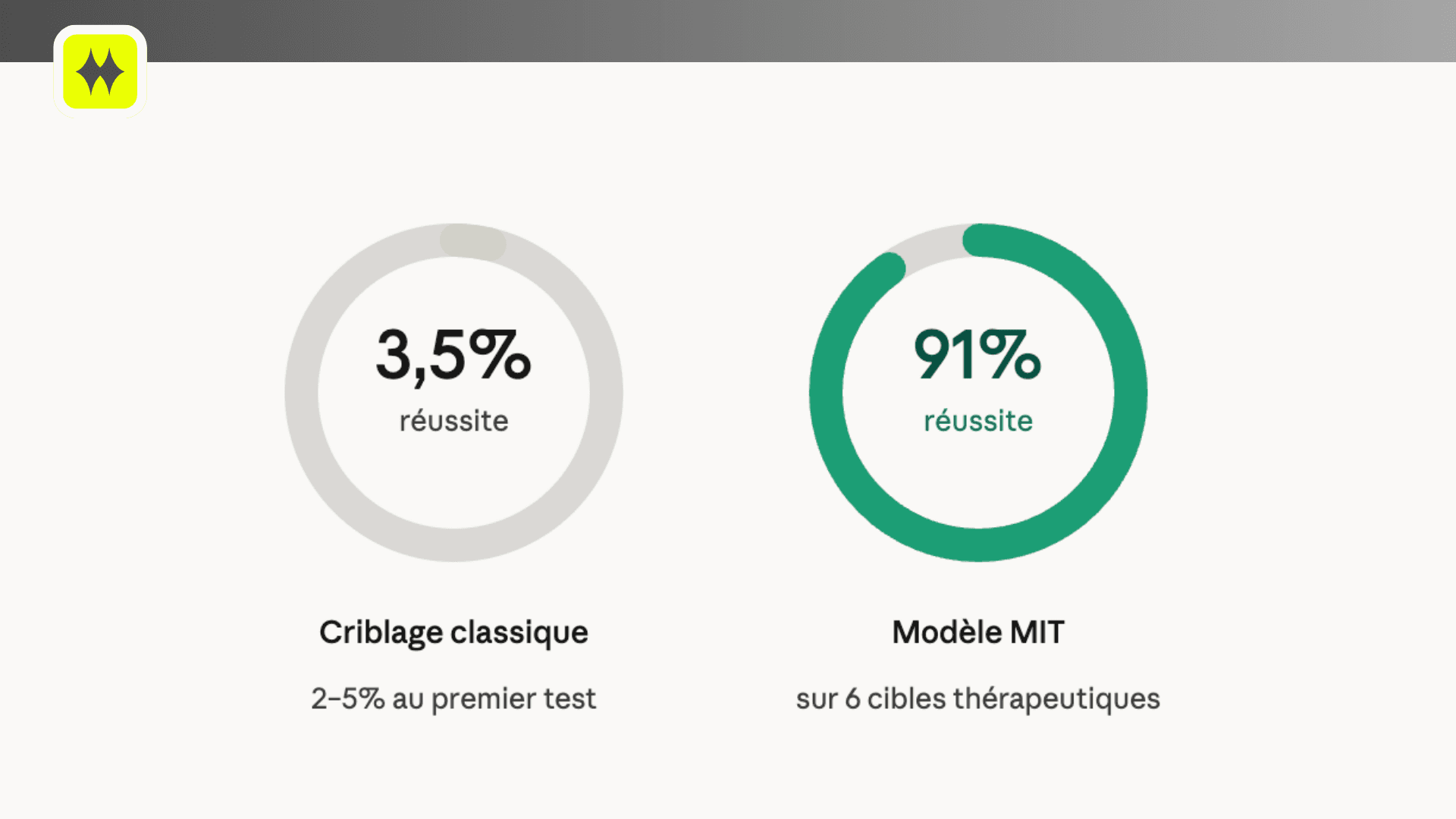
Les équipes ont testé le modèle sur six cibles thérapeutiques : deux liées au cancer colorectal, deux à la leucémie myéloïde aiguë, et deux à des maladies auto-immunes. Dans les six cas, au moins un des dix candidats générés a montré une interaction expérimentalement mesurable. Le taux de succès moyen au premier test est de 91%, contre 2 à 5% avec les méthodes de criblage classiques.
- 91% de corrélation entre simulations et résultats expérimentaux sur 6 cibles
- Temps de génération d'un candidat : 4 heures contre 18 à 24 mois en méthode classique
- Coût de génération par candidat : environ 200 dollars de compute
- Testé sur 6 cibles thérapeutiques dans 3 domaines pathologiques
- Publication dans Nature Biotechnology, mars 2026
- Code et poids du modèle disponibles sous licence Creative Commons
Les partenariats déjà en place
Le laboratoire du MIT a conclu des accords de collaboration avec trois laboratoires pharmaceutiques, dont Novartis et Sanofi. Ces accords permettent aux industriels d'accéder au modèle en échange de données propriétaires sur des interactions protéiques non publiées. C'est un modèle de co-développement qui accélère à la fois la recherche académique et les pipelines industriels.
Ce que ça ne résout pas encore
La prédiction computationnelle reste une approximation. Les effets secondaires, la pharmacocinétique, la toxicité à long terme, les interactions médicamenteuses : aucune de ces variables n'est modélisée de façon fiable. L'IA accélère la phase de découverte, mais les phases 1, 2 et 3 des essais cliniques restent incontournables. C'est là que se joue l'économie du médicament.
Questions fréquentes
Quand ces médicaments seront-ils disponibles pour les patients ?
Les premières molécules générées par le modèle entrent en phase préclinique fin 2026. Les essais cliniques en 3 phases prennent en moyenne 7 à 10 ans. Les premiers médicaments issus de cette approche ne seront pas disponibles avant 2033-2035 au plus tôt.
Le modèle est-il accessible aux chercheurs indépendants ?
Oui. Le code source et les poids du modèle sont disponibles sous licence Creative Commons depuis la publication dans Nature Biotechnology en mars 2026.
AlphaFold et ce modèle du MIT font-ils la même chose ?
Non. AlphaFold prédit la structure 3D d'une protéine à partir de sa séquence. Le modèle du MIT résout le problème inverse : il génère des séquences de protéines qui produiront une interaction spécifique avec une cible thérapeutique donnée.